MDHearingAid offers some of the most affordable, high-quality hearing aids on the online market; but...
Read More









MDHearingAid offers some of the most affordable, high-quality hearing aids on the online market; but...
Read More
Because Market is an online company focused on providing support for older adults who suffer...
Read More
Dermafacs offers a solution to damaged skin with their RecoverX skin repair cream — but...
Read More
FOCL is a CBD line that claims to offer some relief from tension. But does...
Read MoreCurology is a skincare company that provides personalized products based on each customer's skin type,...
Read More
Performer 8s all-natural ingredients seek to help men restore sexual confidence in the bedroom; but...
Read More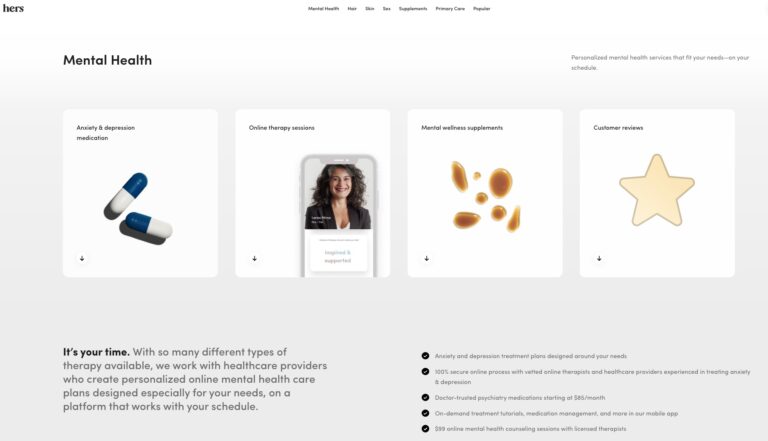
Hers is one of many online mental health services. Read on to find out if...
Read More
To determine if people trust AI for medical and health advice, we surveyed 1,015 participants...
Read More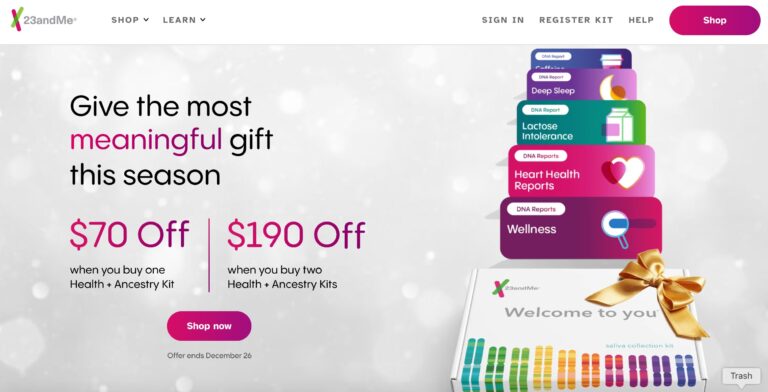
When it comes to consumer genetics testing, 23andMe ranks at the top. Here, we give...
Read MoreTalkspace is a telemedicine provider of mental health services that offers patients on demand virtual...
Read More
When looking for the best online psychiatrist, look for a service that is HIPAA compliant,...
Read More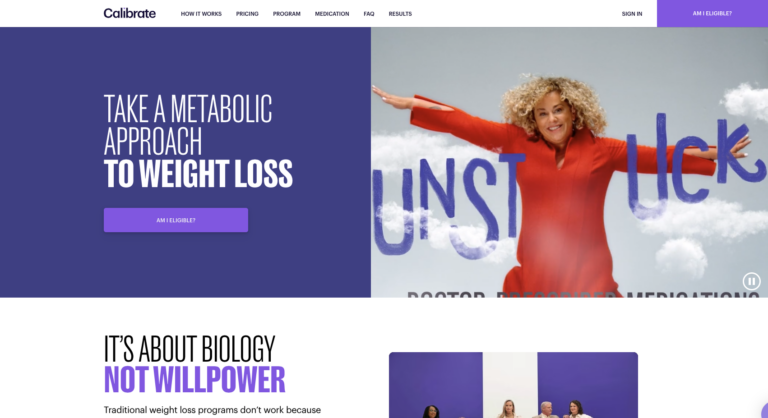
Calibrate is a telehealth company that offers a virtual, year-long weight loss program. This program...
Read More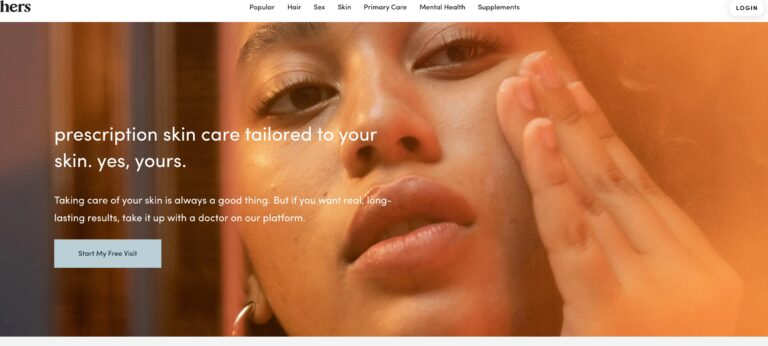
Hers offers prescription skin care creams for women, one for acne and one for anti-aging....
Read More
Eight Sleep offers some of the most advanced smart mattresses money can buy; but are...
Read More
Shouldn’t vaccinations, ambulance rides, and diapers be free? We surveyed 1,000+ Americans to uncover which...
Read More
Have you ever lied to your doctor? If your answer is yes, don’t worry, you’re...
Read More
Let’s take a closer look at how many Americans report using different drugs (including marijuana,...
Read More
We’ve put together a complete review of Hims vs. Roman to help you choose the...
Read More
We’ve compiled a comprehensive Hims review that will tell you everything you need to know...
Read More
In a sea of options, MobileHelp is one medical alert system that stands out to...
Read More
Today, we’re deep diving with a Cerebral review to give you some insider info to...
Read More
Roman is a remote men’s health service that focuses on providing treatments and prescriptions for...
Read MoreBoth BlueChew and Hims can help you save on your ED pills, but comparing the...
Read More
The best online doctor and medical services in 2023; include industry staples like Teladoc and...
Read More
Goop is a lifestyle company seeking to make a splash in the e-commerce retail market...
Read More
BlueChew and Roman both men's health brands that offer erectile dysfunction treatments and ED pills....
Read More
REX MD is a telehealth company that wants to reimagine the world of men’s healthcare....
Read More
The Hims Total Hair Package contains clinically proven components that are effective at stopping hair...
Read More
For years, HairClub's been offering lifelines for those with hair loss, but are their hair...
Read More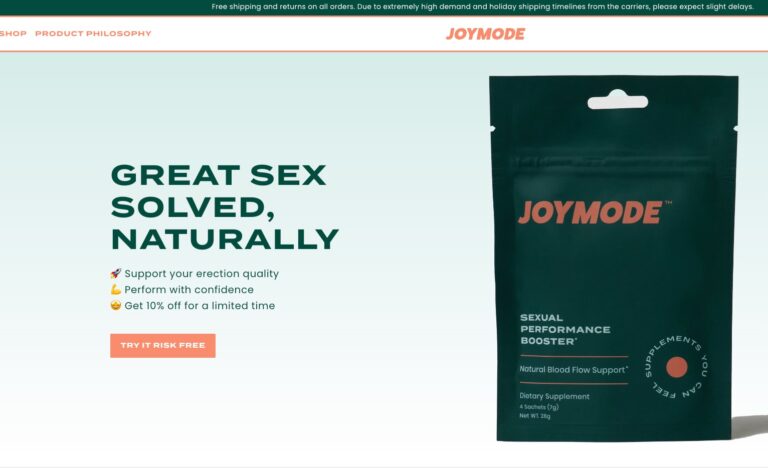
Their two standout products include Men’s Testosterone Support Complex and Sexual Performance Booster. But, in...
Read More
Hers is one telemedicine company looking to provide women with a solution for hair loss....
Read More
Walgreens is one of America's leading pharmacy chains, but does their app meet the expectations...
Read More
Obesity affects over 70% of the population. Learn how Found Health, Inc. is changing some...
Read More
K Health is shaping the future of healthcare with its affordable pricing and innovative approach;...
Read More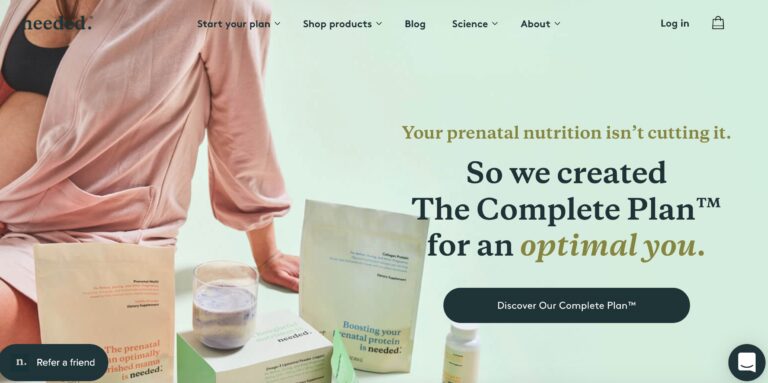
Needed is a supplement brand aimed at providing nutrients for pregnant mothers. But do you...
Read More
Nava MD offers personalized skin care treatment plans for several skin conditions, from acne to...
Read More
Everlywell is a telehealth company that offers affordable at-home testing kits for tons of health...
Read More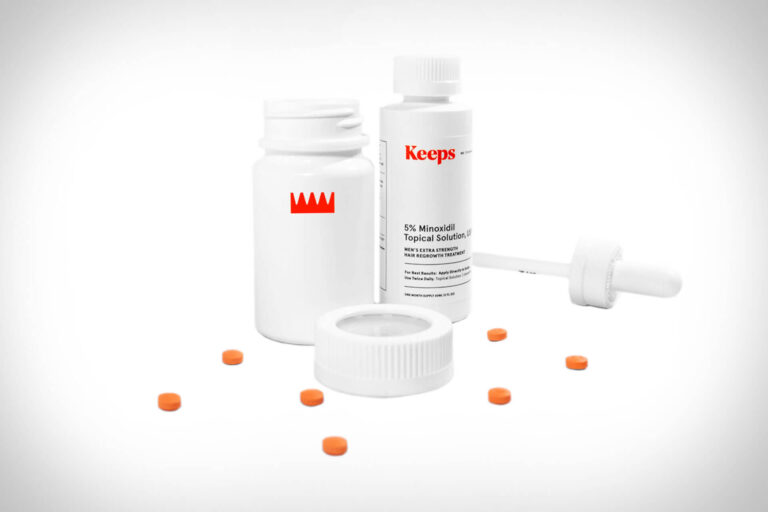
The telehealth provider Keeps offers clinically proven FDA-approved hair-loss treatment for men. In this review,...
Read More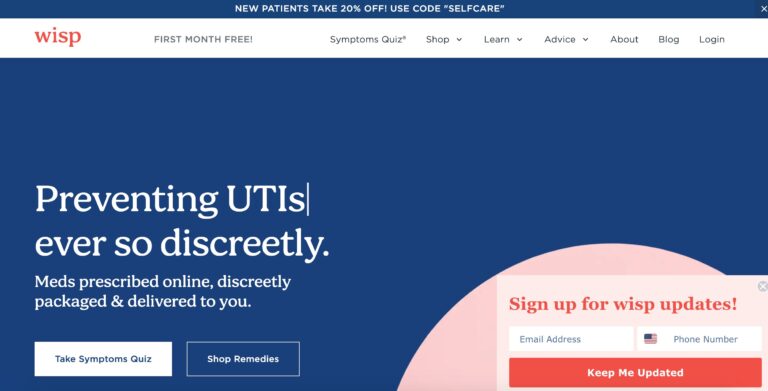
Wisp is a company that offers direct to consumer healthcare focusing on reproductive and sexual...
Read More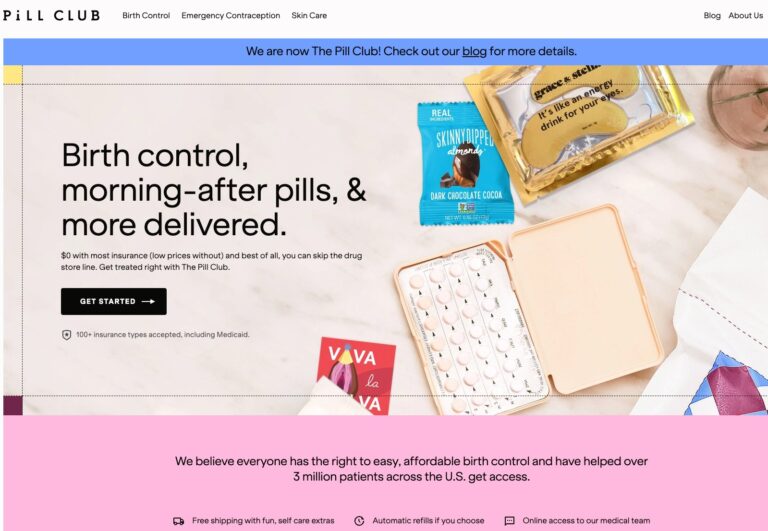
The Pill Club is a birth control provider that seeks to remove barriers to womens...
Read More

Exploring Pharmacy Density Nationwide
Read More
We’ve surveyed over 1,000 people to get their takes on this latest political controversy surrounding...
Read More
Let’s take a closer look at how many Americans report using different drugs (including marijuana,...
Read More
Have you ever lied to your doctor? If your answer is yes, don’t worry, you’re...
Read More
Shouldn’t vaccinations, ambulance rides, and diapers be free? We surveyed 1,000+ Americans to uncover which...
Read More
To determine if people trust AI for medical and health advice, we surveyed 1,015 participants...
Read More